FASTA Files
- Aadit Mathur

- Jun 25
- 2 min read
Summary
FASTA files are a format providing a simple way of storing nucleotide or amino acid sequences. They have a single-line header at the top that starts with ">", followed by several lines of the sequence. Each header marks the beginning of a new sequence.
To read sequences from FASTA files, simply read all the lines after the header and until you encounter a header once more (code example at the bottom).
FASTA Files
FASTA files are a widely used format for storing nucleotide and amino acid sequences. The files usually end in .fasta, .fas, or .fa. Sometimes, to specify nucleotide sequences, .fna is used instead, and for amino acids, .faa is used. The format is text-based, making it easy to use. Just open the file in a text editor to easily read the sequences.
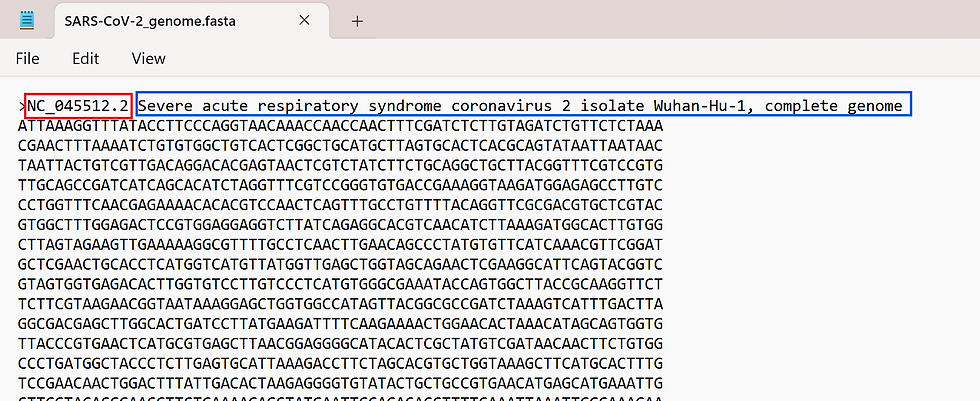
The first line of the file is the header, and it contains information about where the data came from. It starts with ">" and is followed by various bits of data. In this case, the line starts with the accession number (unique ID) of the sequence, followed by the name of the sequence. Other metadata can be added in this line.
Oftentimes, what we care about are the following lines, which contain the actual sequence. The FASTA format doesn't specify a line length limit, but 80 characters or less is often recommended. In the SARS-CoV-2 genome above, each line is only 70 characters.
To parse the file, we can search for the header line and then keep reading lines (until we get to another header, which would be the start of another sequence).
Below is an example written in Python.
Comments